Deep Learning and Human Intelligence – Part 1 of 2
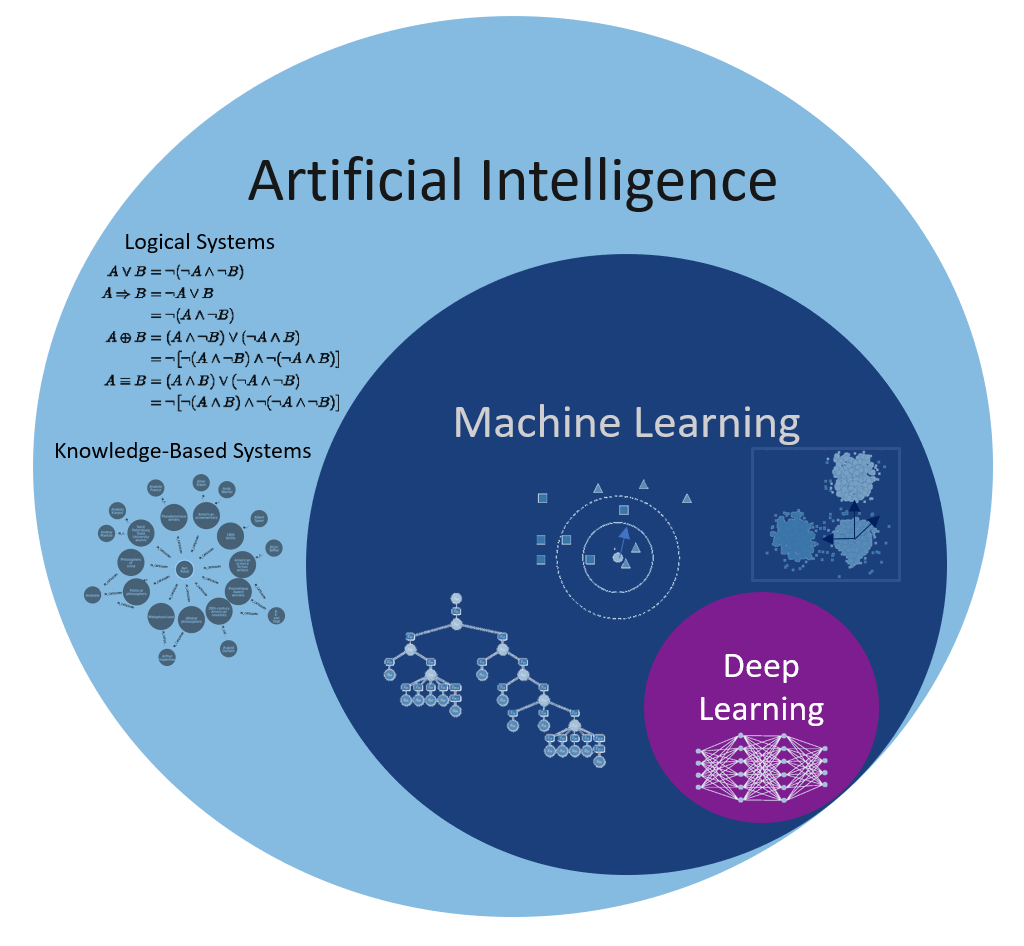
Many people are under the impression that the new wave of data science, machine learning and/or digitalization is new, that it did not exist before. But its history is as long as the history of humanity and/or science itself. The scientific discovery could hardly take place without the necessary data. Even the process of discovering the numbers included elements of machine learning: pattern recognition, comparison between different groups (ranking), clustering, etc. So what differentiates mathematical formulas from machine learning and how does it relate to artificial intelligence?
There is no difference between the two if seen from the perspective of formulas however, such a perspective limits the type of data to which they can be applied. Data stored via tables consist of structured data and are stored in so-called relational databases. The reason for such a data storage is the connection between different fields that assume a well-established structure in advance, such as a company’s sales or balance sheet. However, with the emergence of personal computers, many of the daily activities have been digitalized: music, pictures, movies, and so on. All this information is stored unrelated to other data and therefore called unstructured data.
Copyright: IEEE International Conference on Computer Vision (ICCV), 2015, DOI: 10.1109/ICCV.2015.428
The essence of scientific discoveries was and will be structure. Not surprisingly, the mathematical formulas revolve around relations between variables – information, in general. For example, Galileo derived the law of falling balls from measuring the successive hight of a falling ball. The main difficulty was to obtain measurements at regular time intervals. What about if the data is not structured, which mathematical formula should be applied then? There is a distribution of people’s height, but no distribution for the pictures taken in all holidays for the last year, there is an amplitude for acoustic signals, but no function that detects the similarity between two songs. This is one of the reasons why machine learning focuses heavily on clustering and classification.
Roughly speaking, these simple examples are enough to categorize the difference between scientific discovery and machine learning. Science is about discovering relationships between different variables, Machine Learning tries to automatize processes. Every technical improvement is part of the automation, so why is everything different in this case? Because the current automation deals with human intelligence. The car automates the walking, the kitchen stove the fire, but Machine Learning parts of the human intelligence. There is a difference between the previous automation steps and those of human intelligence. All the previous ones are either outside the human body – such as Fire – or unconsciously executed (once learned) – walking, spinning, etc. The automation induced by Machine Learning affects a part of the human intelligence that we consciously perceive. Of course, today’s machine learning tools are unable to automate all human intelligence, but it is a fascinating step in that direction.
A breakthrough in Machine Learning tasks was achieved in 2012 when the first Deep Learning algorithm for detecting types of images, reached near-human accuracy. It could appreciate the likelihood that the image is a human face, a train, a ball or a fish without having “seen” the picture before. Such an algorithm can be used in various areas: personally – facial recognition in pictures and/or social media – as tagging of images or videos, medicine – cancer detection, etc. For understanding such cutting-edge issues of classification, one cannot avoid understanding how Deep Learning works. To see the beauty of such algorithms and, at the same time, to be able to comprehend the difficulty of working with them, an example will be the best guide.
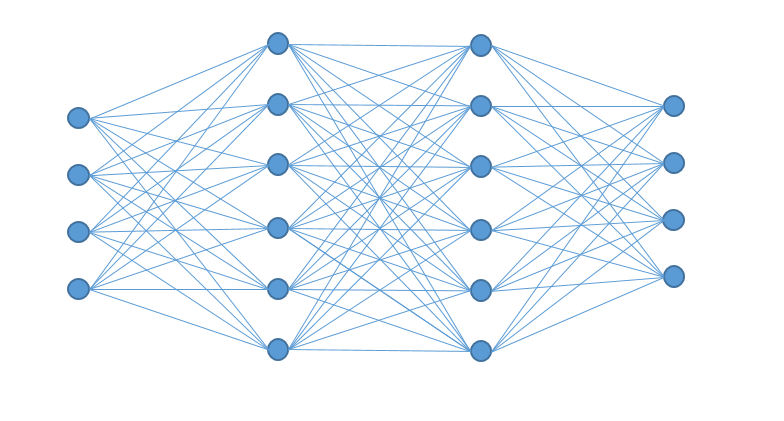
The building blocks of Deep Learning are neurons, operational units, which perform mathematical operations or logical operations like AND, OR, etc., and are modelled after the neurons in the brain. Already in the 1950’s two neuroscientist, Hubel and Wiesel, observed that not all neurons in the brain are responding in the same fashion to visual stimuli. Some responded only to horizontal lines, whereas others to vertical lines, with other words, the brain is constructed with specialized neurons. Groups of such neurons are called, in the Machine Learning community, layers. Like in the brain, neurons with different properties are clustered in different layers. This implies that layers have also specific properties and have to be arranged in a specific way, called architecture. It is this architecture which differentiates Deep Learning from Artificial Neuronal Networks (ANN are similar to a layer).
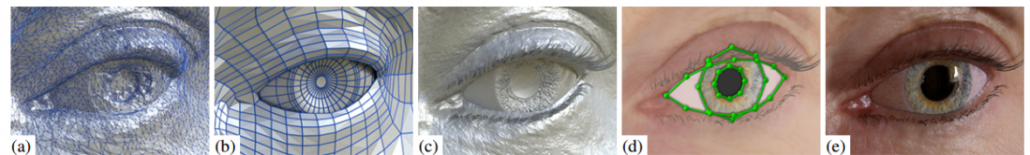
Unfortunately, scientists still haven’t figured out how the brain works, thus to discover how to train Deep Learning from data was not an easy task, and is also the reason why another example is used to explain the training of Deep Learning: the eye. One has always to remember: once it is known how Deep Learning works, it is simple to find example which illustrates the working mechanism. For such an analogy, it is sufficient for someone without any knowledge about Deep Learning, to keep in mind only the elements that compose such architectures: input data, different layers of neurons, output layers, ReLu’s.
Input data are any type of information, in our example it is light. Of course, that Deep Learning is not limited only to images or videos, but also to sound and/or time series, which would imply that the example would be the ear and sound waves, or the brain and numbers.
Layers can be seen as cells in the eye. It is well known that the eye is formed of different layers connected to each other with each of them having different properties, functionalities. The same is true also for the layers of a Deep Learning architecture: one can see the neurons as cells of the layer as the tissue. While, mathematically, the neurons are nothing more than simple operations, usually linear weight functions, they can be seen as the properties of individual cells. Each layer has one weight matrix, which gives the neuron (and layer) specific properties depending on the data and the task at hand.
It is here that the architecture becomes very important. What Deep Learning offers is a default setting of the layers with unknown weights. One can see this as trying to build an eye knowing that there are different types of cells and different ways how tissues of such cells can be arranged, but not which cell exactly is needed (with what properties) and which arrangement of layers works best. Such an approach has the advantage that one is capable of building any type of organ desired, but the disadvantage is also very obvious: it is time consuming to find the appropriate cell properties and layers arrangements.
Still, the strategy of Deep Learning is a significant departure from the Machine Learning approaches. The performance of Machine Learning methods is as good as the features engineering performed by Data Scientists, and thus depending on the creativity of the Data Scientist. In the case of Deep Learning the engineers of the features is performed automatically as part of the model building. This is a huge improvement, as the only difficult task is to have enough data and computer power to find the right weights matrices. Such an endeavor was performed also by nature for the eye — and is also the reason why one can choose it as an example for Deep Learning — evolution. It is not surprising that Deep Learning is one of the best direction scientists have of Artificial Intelligence today.
The evolution of the eye can be seen, from the perspective of Data Scientists, as the continuous training of a Deep Learning architecture which enables to recognize and track one or more objects. The performance of the evolutional process can be summed up as the fine tuning of the cells which are getting more and more susceptible to light and the adaptation of layers to enable a better vision. Different animals in different environments and different targets — as the hawk and the fly — developed different eyes than humans, but they all work according to the same principle. The tasks that Deep Learning is performing today are similar, for example it can be used to drive cars but there is still a difference: there is no connection to other organs. Deep Learning is not the approximation of an Artificial Organism, like an android, but a simplified Artificial Organ that can work on its own.
Returning to the working mechanism of the Deep Learning architecture, we can already follow the analogy of what happens if a ray of light is hitting the eye. Once the eye is fully adapted to the task, one can followed how the information enters the Deep Learning architecture (Artificial Eye) by penetrating the input layer. already here arises the question, what kind of eye is the best? One where a small source of light can reach as many neurons as possible, or the one where the light sources reaches only few neurons? In order to take such a decision, a last piece of the puzzle is required: ReLu. One can see them as synapses between neurons (cells) and/or similarly for tissue. By using continuous functions, such as the shape of the latter ‘S’ (called sigmoid), the information from one neuron will be distributed over a large number of other neurons. If one uses the maximum function, then only few neurons are updated with processed information from earlier layers.
Such sparse structures between neurons, was a major improvement in the development of the technique of training Deep Learning architectures. Again, it has a strong evolutionary analogy: energy efficiency. By needing less neurons, the tissues and architecture are both kept to a minimal size which enables flexibility in development and less energy. As the information is process by the different layers, the Artificial Eye is gathering more and more complex (non-linear) structures — the adapted features –, which help to decide, from past experience, what kind of object is detected.
This was part 1 of 2 of the article series. Continue with Part 2.








 kann allgemein mit folgender Formel bestimmt werden
kann allgemein mit folgender Formel bestimmt werden  .
. aus dem Datenraum
aus dem Datenraum  , die ein Trainer dem Lernverfahren vorgibt, um Zielkonzept c zu erlernen, eine Hypothese aus dem Hypothesenraum
, die ein Trainer dem Lernverfahren vorgibt, um Zielkonzept c zu erlernen, eine Hypothese aus dem Hypothesenraum  des Lernverfahrens zu ermitteln, welche (möglichst) alle positiven Beispiel
des Lernverfahrens zu ermitteln, welche (möglichst) alle positiven Beispiel  umfasst und (möglichst) alle negativen Beispiele
umfasst und (möglichst) alle negativen Beispiele  ausschließt.
ausschließt.
 .
. und im Fall von Dummy-Encoding
und im Fall von Dummy-Encoding  neue Bool’sche Eigenschaften.
neue Bool’sche Eigenschaften. und für allgemeine endliche Eigenschaften
und für allgemeine endliche Eigenschaften . Diese Repräsentation ist sehr eingeschränkt und erlaubt es nur einzelne und keine kombinierten Konzepte zu erlernen. Sie ist daher eigentlich nur von theoretischem Interesse und wird – soweit bekannt – in keinem praktisch eingesetzten Lernverfahren genutzt.
. Diese Repräsentation ist sehr eingeschränkt und erlaubt es nur einzelne und keine kombinierten Konzepte zu erlernen. Sie ist daher eigentlich nur von theoretischem Interesse und wird – soweit bekannt – in keinem praktisch eingesetzten Lernverfahren genutzt. . Mit dieser Sprache können alle Eigenschaften zwar separat auf beliebige Teilmengen generalisiert werden, Korrelationen zwischen Eigenschaften werden jedoch nicht berücksichtigt.
. Mit dieser Sprache können alle Eigenschaften zwar separat auf beliebige Teilmengen generalisiert werden, Korrelationen zwischen Eigenschaften werden jedoch nicht berücksichtigt. . Auf beliebige endliche Eigenschaften übertragen, kann diese Aussage zu
. Auf beliebige endliche Eigenschaften übertragen, kann diese Aussage zu  verallgemeinert werden.
verallgemeinert werden.

 die ein „konsistenter Lernalgorithmus“
die ein „konsistenter Lernalgorithmus“ und einer Unsicherheit
und einer Unsicherheit  (bzw. einer Wahrscheinlichkeit von
(bzw. einer Wahrscheinlichkeit von  ) zu erlernen, abgeschätzt werden mit
) zu erlernen, abgeschätzt werden mit![\[m \geq \frac{1}{\epsilon}(ln{(|H|)} + ln{(\frac{1}{\delta})})\]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-901f897bb0040e542c0eeaa14098b677_l3.png "Rendered by QuickLaTeX.com")
 Mrd. Punkten. Mit einer einfachen bool’schen Kodierung ergibt sich
Mrd. Punkten. Mit einer einfachen bool’schen Kodierung ergibt sich  und
und  .
. – wie auch immer – klassifizieren, so würden wir für den Einsatz von Naive Bayes oder unbegrenzten DecisionTrees mindestens 76.145 Datensätze benötigen. Weder die monatlichen Daten von Produkt A noch Produkt B würden ausreichen.
– wie auch immer – klassifizieren, so würden wir für den Einsatz von Naive Bayes oder unbegrenzten DecisionTrees mindestens 76.145 Datensätze benötigen. Weder die monatlichen Daten von Produkt A noch Produkt B würden ausreichen. kann etwas außerhalb des Hypothesenraums liegen, der durch das eingesetzte Lernverfahren erfasst wird. Dies bedeutet, dass wir im Hypothesenraum des Lernverfahrens nur eine Näherung
kann etwas außerhalb des Hypothesenraums liegen, der durch das eingesetzte Lernverfahren erfasst wird. Dies bedeutet, dass wir im Hypothesenraum des Lernverfahrens nur eine Näherung  erlernen können, die möglichst gut sein sollte. Solch ein – als agnostisch bezeichnetes – Lernverfahren muss daher bestrebt sein den Fehler zwischen den Trainingsdaten und dem Fehler der sich durch das Erlernen der Näherung
erlernen können, die möglichst gut sein sollte. Solch ein – als agnostisch bezeichnetes – Lernverfahren muss daher bestrebt sein den Fehler zwischen den Trainingsdaten und dem Fehler der sich durch das Erlernen der Näherung ![\[m \geq \frac{1}{2\epsilon^2}(ln{(|H|)} + ln{(\frac{2}{\delta})})\]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-0630d9d4a9a075de904ef5d5610c93eb_l3.png "Rendered by QuickLaTeX.com")





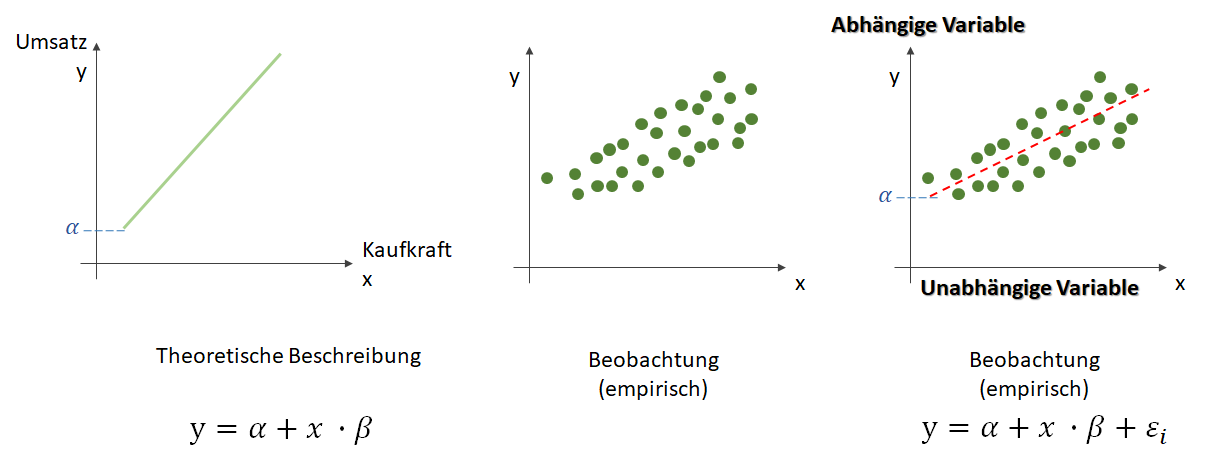
 , die unsere Punktwolke – mit der wir uns zutrauen, Vorhersagen über die abhängige Variable vornehmen zu können – möglichst gut beschreibt. Dabei ist
, die unsere Punktwolke – mit der wir uns zutrauen, Vorhersagen über die abhängige Variable vornehmen zu können – möglichst gut beschreibt. Dabei ist  der Zielwert (abhängige Variable) und
der Zielwert (abhängige Variable) und  der Eingabewert. Wir arbeiten also in einer zwei-dimensionalen Welt. Variablen, die die Funktion mathematisch definieren, werden oft als griechische Buchstaben darsgestellt. Die Variable
der Eingabewert. Wir arbeiten also in einer zwei-dimensionalen Welt. Variablen, die die Funktion mathematisch definieren, werden oft als griechische Buchstaben darsgestellt. Die Variable  (Alpha) ist der
(Alpha) ist der  . Dieser wird als Bias, selten auch als Default-Wert, bezeichnet. Der Bias ist also der Wert, wenn die
. Dieser wird als Bias, selten auch als Default-Wert, bezeichnet. Der Bias ist also der Wert, wenn die  (Beta) beschreibt die Steigung.
(Beta) beschreibt die Steigung. ein Fehler
ein Fehler  existiert. Diesen Fehler wollen wir in diesem Artikel ignorieren.
existiert. Diesen Fehler wollen wir in diesem Artikel ignorieren. statt
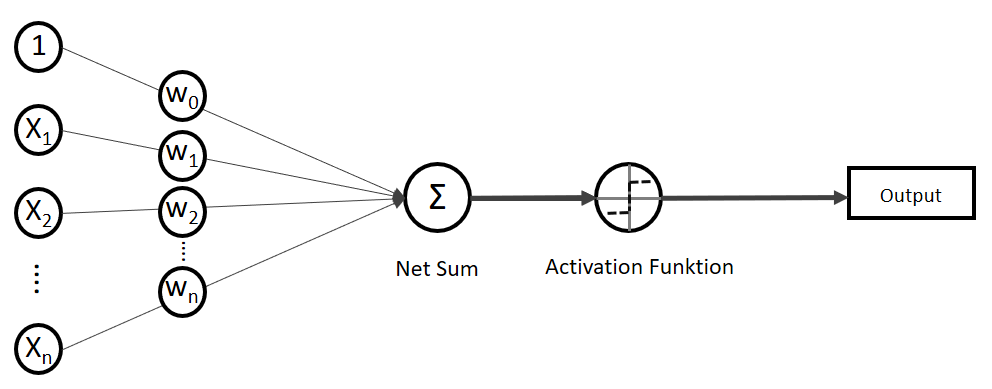
statt  ) sind nichts anderes als Gewichtungen zwischen den Eingaben.
) sind nichts anderes als Gewichtungen zwischen den Eingaben.![\[y = w_{0} \cdot x_{0} + w_{1} \cdot x_{1} + \ldots + w_{n} \cdot x_{n}\]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-19728ae5f9e3065a4c17d50c417e0a8b_l3.png "Rendered by QuickLaTeX.com")
 . Verkürzt ausgedrückt:
. Verkürzt ausgedrückt:![\[y = \sum_{i=0}^n w_{i} \cdot x_{i}\]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-3bbcbb30d482e7a1fd8da528030d6561_l3.png "Rendered by QuickLaTeX.com")
![\[y = w^T \cdot x\]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-939f4bae6617db519d13b502d6337ee1_l3.png "Rendered by QuickLaTeX.com")
 .
.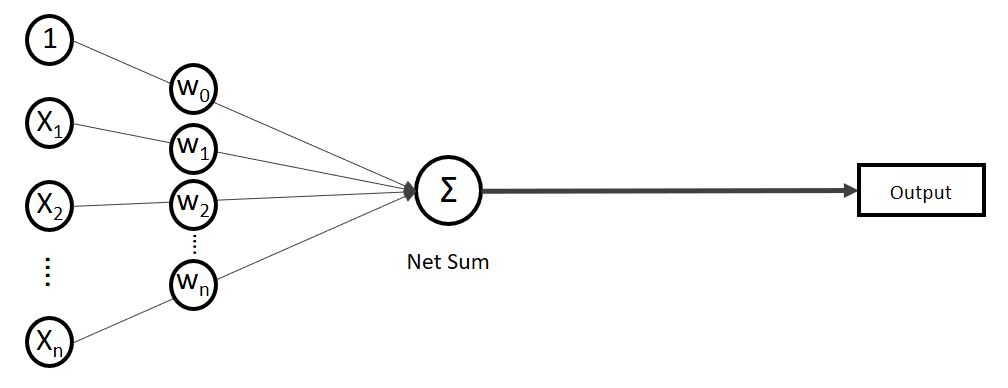
 . Auf der linken Seite finden wir alle Eingabewerte, wobei der erste Wert statisch mit 1.0 belegt ist, nur für den Zweck, den Bias (
. Auf der linken Seite finden wir alle Eingabewerte, wobei der erste Wert statisch mit 1.0 belegt ist, nur für den Zweck, den Bias (
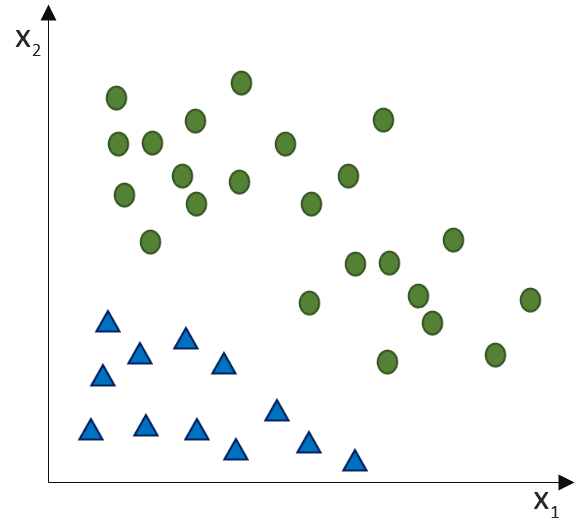
 (Phi), die uns die stetigen Werte der Nettoeingabe in einen binären Wert (z. B. 0 oder 1) umwandelt.
(Phi), die uns die stetigen Werte der Nettoeingabe in einen binären Wert (z. B. 0 oder 1) umwandelt. 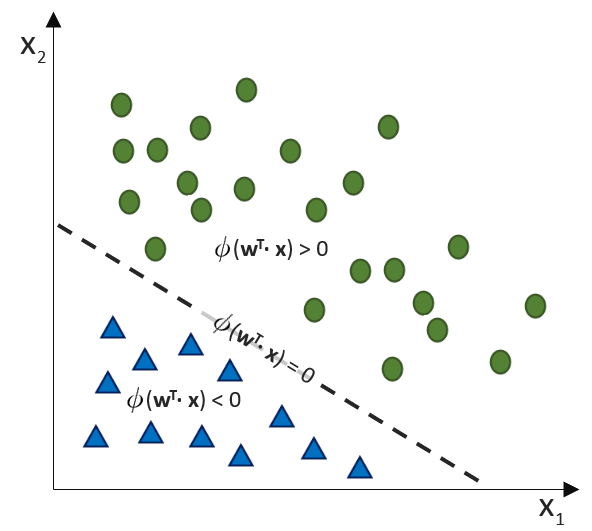
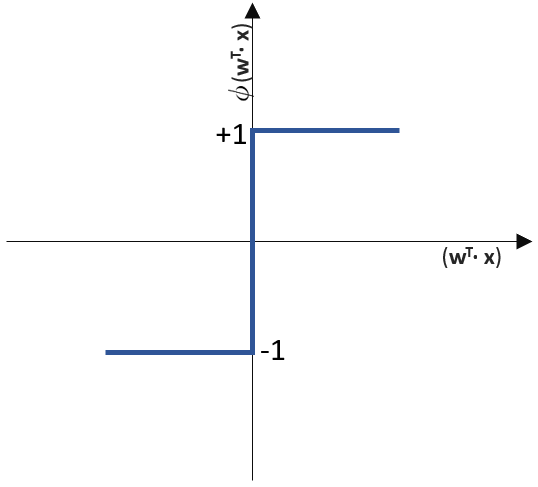
![\[ y = \phi(w^T \cdot x) = \left\{ \begin{array}{12} 1 & w^T \cdot x > 0\\ -1 & \text{otherwise} \end{array} \]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-7b828cf4bbabf9e1a84ec9b628d51249_l3.png "Rendered by QuickLaTeX.com")
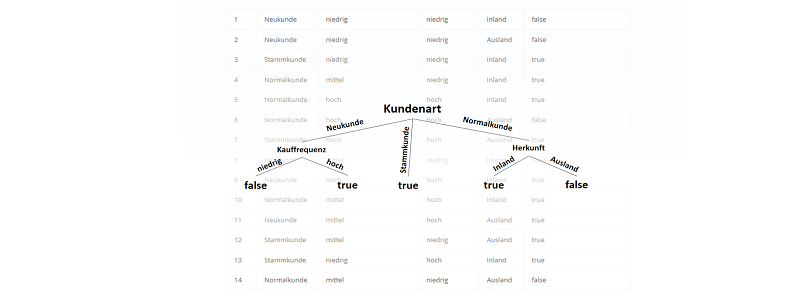
 ) im Sinne des ID3-Algorithmus ist die Differenz aus der Entropie (
) im Sinne des ID3-Algorithmus ist die Differenz aus der Entropie ( ) (siehe Teil 1 der Artikelserie
) (siehe Teil 1 der Artikelserie  ), der im Attribut vorkommt:
), der im Attribut vorkommt:

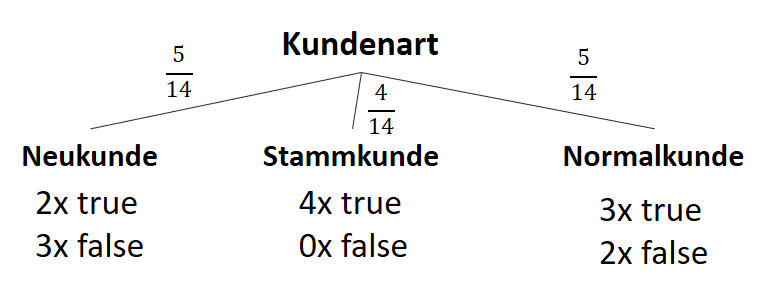



![\[ IG(S, A_{Kundenart}) = - \sum_{i=1}^n \frac{\bigl|S_i\bigl|}{\bigl|S\bigl|} \cdot H(S_i) \]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-6380891cc61bb855d4208480091fdef0_l3.png "Rendered by QuickLaTeX.com")
![\[ IG(S, A_{Kundenart}) = H(S) - \frac{\bigl|S_{Neukunde}\bigl|}{\bigl|S\bigl|} \cdot H(S_{Neukunde}) - \frac{\bigl|S_{Stammkunde}\bigl|}{\bigl|S\bigl|} \cdot H(S_{Stammkunde}) - \frac{\bigl|S_{Normalkunde}\bigl|}{\bigl|S\bigl|} \cdot H(S_{Normalkunde}) \]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-b3c9d40c7a171fd7b9e5fbed0aed041a_l3.png "Rendered by QuickLaTeX.com")
![\[ IG(S, A_{Kundenart}) = 0.94 - \frac{5}{14} \cdot 0.97 - \frac{4}{14} \cdot 0.00 - \frac{5}{14} \cdot 0.97 = 0.247 \]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-33144ba5df6858348666daef5a5cf5d3_l3.png "Rendered by QuickLaTeX.com")
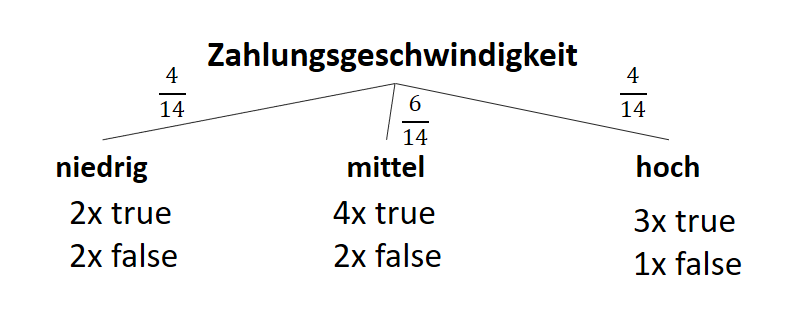



![\[ IG(S, A_{Zahlungsgeschwindigkeit}) = H(S) - \frac{\bigl|S_{niedrig}\bigl|}{\bigl|S\bigl|} \cdot H(S_{niedrig}) - \frac{\bigl|S_{mittel}\bigl|}{\bigl|S\bigl|} \cdot H(S_{mittel}) - \frac{\bigl|S_{schnell}\bigl|}{\bigl|S\bigl|} \cdot H(S_{schnell}) \]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-a80e61c0adf9dff8ca219a28bce359cd_l3.png "Rendered by QuickLaTeX.com")
![\[ IG(S, A_{Zahlungsgeschwindigkeit}) = 0.94 - \frac{4}{14} \cdot 1.00 - \frac{6}{14} \cdot 0.92 - \frac{4}{14} \cdot 0.81 = 0.029 \]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-ed4f1d54de66a9956bc2c1e53a0de2e8_l3.png "Rendered by QuickLaTeX.com")



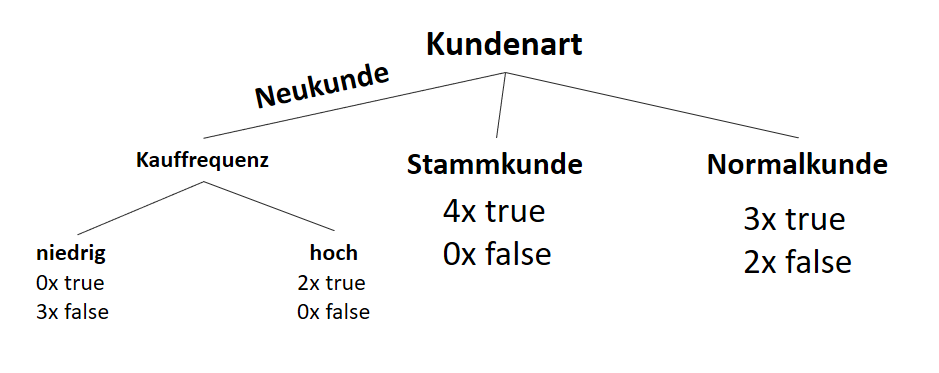
![\[ IG(S, A_{Kauffrequenz}) = H(S) - \frac{\bigl|S_{niedrig}\bigl|}{\bigl|S\bigl|} \cdot H(S_{niedrig}) - \frac{\bigl|S_{hoch}\bigl|}{\bigl|S\bigl|} \cdot H(S_{hoch}) \]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-cbcfb6a5b118cfc2df4185ef03e4e1dd_l3.png "Rendered by QuickLaTeX.com")
![\[ IG(S, A_{Kauffrequenz}) = 0.94 - \frac{7}{14} \cdot 1.00 - \frac{7}{14} \cdot 0.59 = 0.150 \]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-dc9f358f2d2cd071b815b3f6c571f813_l3.png "Rendered by QuickLaTeX.com")
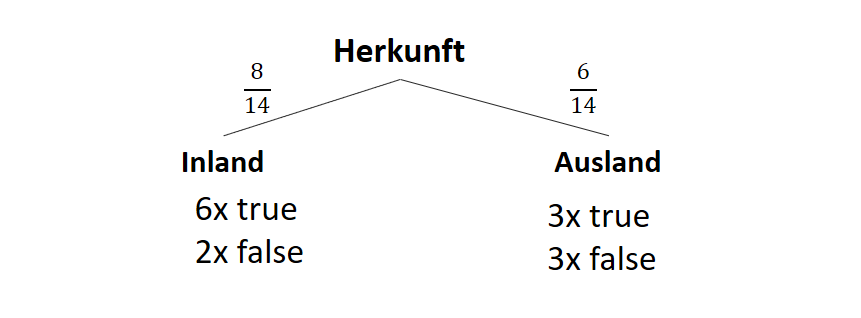


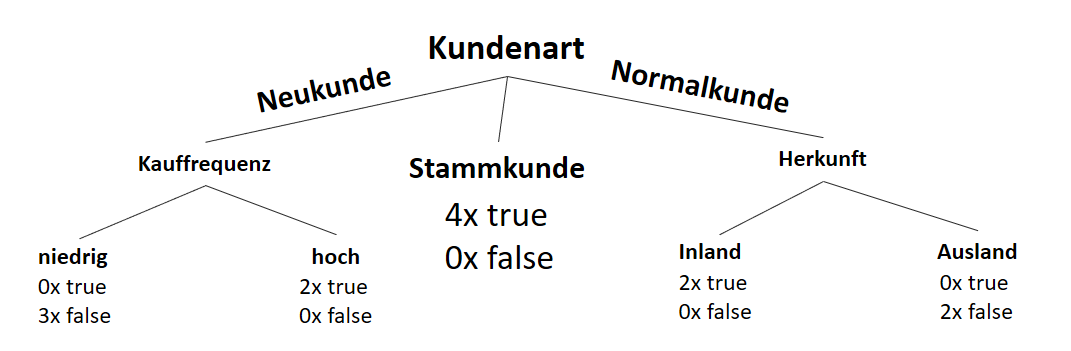
![\[ IG(S, A_{Herkunft}) = H(S) - \frac{\bigl|S_{Inland}\bigl|}{\bigl|S\bigl|} \cdot H(S_{Inland}) - \frac{\bigl|S_{Ausland}\bigl|}{\bigl|S\bigl|} \cdot H(S_{Ausland}) \]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-3a0ecd4412fa44ca26c64ea16aa536d1_l3.png "Rendered by QuickLaTeX.com")
![\[ IG(S, A_{Herkunft}) = 0.94 - \frac{8}{14} \cdot 0.81 - \frac{6}{14} \cdot 1.00 = 0.05 \]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-ab4db275a73c092813d00f26ef4c1183_l3.png "Rendered by QuickLaTeX.com")
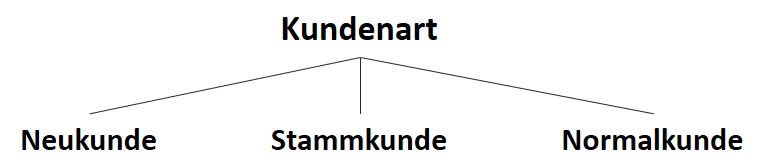
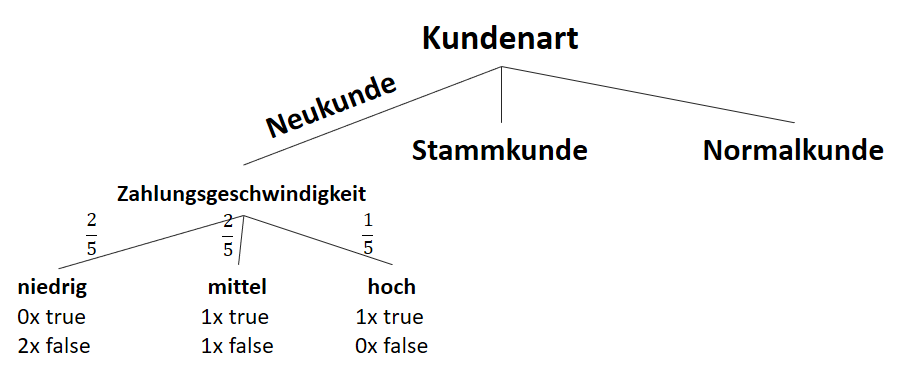



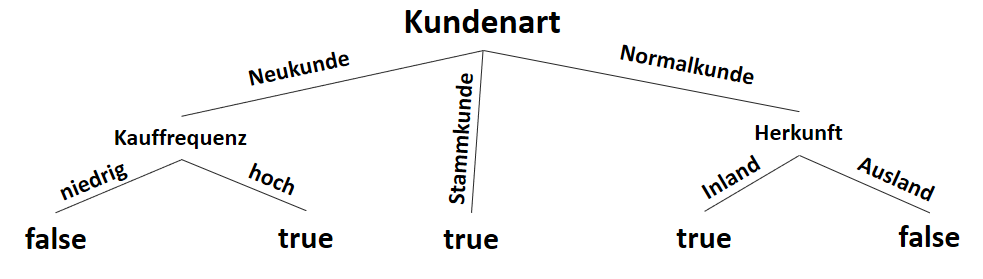
![\[ IG(S_{Neukunde},A_{Zahlungsgeschwindigkeit}) = 0.97 - \frac{3}{5} \cdot 0.00 - \frac{2}{5} \cdot 1.00 - \frac{1}{5} \cdot 0.00 = 0.57 \]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-35c0216b5e69ee73831980225531ecc7_l3.png "Rendered by QuickLaTeX.com")
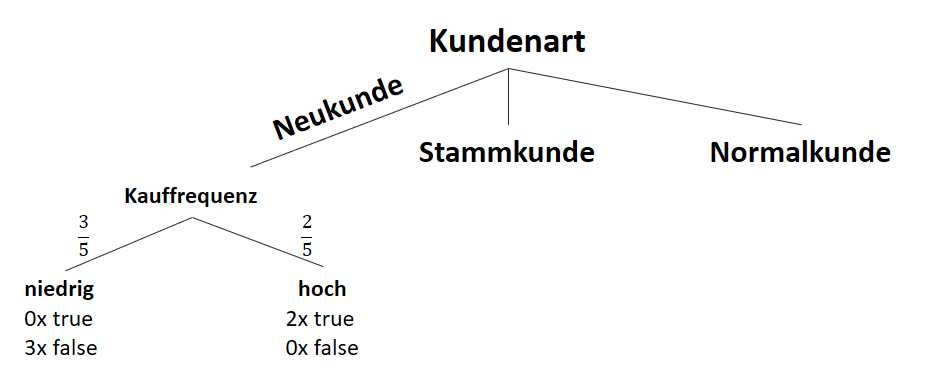
![\[ IG(S_{Neukunde},A_{Kauffrequenz}) = 0.97 - \frac{3}{5} \cdot 0.00 - \frac{2}{5} \cdot 0.00 = 0.97 \]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-5f6849e7156c9505f53b484a8e7f505a_l3.png "Rendered by QuickLaTeX.com")



![\[ IG(S_{Neukunde},A_{Herkunft}) = 0.97 - \frac{3}{5} \cdot 0.92 - \frac{2}{5} \cdot 1.00 = 0.018 \]](http://datasciencehack.com/wp-content/ql-cache/quicklatex.com-890f306c397d0ac28cbbd9a316eb049b_l3.png "Rendered by QuickLaTeX.com")